
博客
利用 Oracles 和实验反馈指导生成式分子设计

AI 生成化学有可能彻底改变科学家在药物研发、健康以及材料科学和工程领域的工作方式。研究人员无需借助“化学直觉”手动设计分子或筛选数百万种现有化学物质,而是可以训练神经网络来提出适合所需特性的新型分子结构。这种能力开辟了广阔的化学空间,这是以前无法系统探索的。
虽然一些早期成功表明,生成式 AI 有望通过提出化学家可能没有考虑过的创造性解决方案来加速创新,但这些成功只是一个开始。生成式 AI 还不是分子设计的灵丹妙药,将 AI 建议的分子转化为现实世界通常比 一些标题所显示的困难得多 。
虚拟设计与现实世界影响之间的差距是当今 AI 驱动的分子设计面临的核心挑战。计算生成式化学模型需要实验反馈和分子模拟来确认其设计的分子是否稳定、可合成且具有功能性。与自驾驶汽车一样,AI 必须经过真实驾驶数据或高保真模拟的训练和验证,才能在不可预测的道路上行驶。正如用于生成式虚拟筛选的 NVIDIA BioNeMo Blueprint 的最新更新所示,如果没有这种基础,理论上两个 AI 系统都有可能生成有前景的解决方案,但在现实世界中进行测试时却失败了。
将 AI 设计与现实联系起来的一种强大方法是通过 oracle (也称为 scoring function) 。在生成式分子设计中,oracle 是一种反馈机制,即一种测试或评估机制,用于告知我们所提议的分子在预期结果方面的表现,通常是分子或实验属性 (例如 potency、safety 和 feasibility) 。
此预言机可以是:
基于实验:用于测量 AI 设计的药物分子与目标蛋白质结合的程度 。
| 实验性 Oracle 类型 | 优势 | 限制 | 实际应用 |
| 体外分析 (例如生化、细胞测试、高通量筛选) | 生物相关性高,小批量快速,可通过自动化实现扩展。 | 成本高昂,吞吐量低于模拟,可能无法捕获 体内效果 。 | 用于在临床试验前识别和优化候选药物的标准。 |
| In vivo 模型 (动物测试) | 提供有关安全性配置文件、剂量等的见解,这些信息通常用于药物审批。 | 成本高昂、速度缓慢、伦理道德方面的问题以及物种差异可能会限制与人类的相关性。 | 用于临床前药物开发,同时越来越多地通过模拟进行补充。 |
这种预言机基于计算,使用高质量计算 (例如分子动力学模拟) 准确预测属性,例如用于计算结合能 (药物放入酶口袋的强度) 的自由能方法,或对材料稳定性的量子化学计算 。当实验室测试速度缓慢、成本高昂或需要进行大规模评估时,这些都是计算机模拟的备用模型。
| 计算 Oracle 类型 | 优势 | 限制 | 实际应用 |
| 基于规则的过滤器 (Lipinski 的 Rule of 5、PAINS 警报等) | 快速标记较差的候选药物,这是广泛接受的启发式算法(heuristics)。 | 过于简单化,可能会拒绝可行的药物 | 用于在药物设计初期快速过滤掉不合适的化合物。 |
| QSAR (从结构中预测活动的统计模型) | 快速、经济高效,适用于 ADMET 属性筛选。 | 需要实验数据,难以处理新的化学物质(novel chemistries)。 | 用于 lead 优化和筛选出较差的候选项。 |
| 分子对接 (基于结构的虚拟筛选) | 快速筛选大型库,说明分子如何与目标结合。 | 与实验结果相比,通常不准确的假设是刚性结构。 | 在早期药物发现中很常见,用于入围前景良好的化合物。 |
| 分子动力学和自由能模拟 (模拟分子随时间变化的行为) | 与对接相比,对灵活性和交互进行建模更真实。 | 计算密集、速度缓慢,需要专业知识。 | 用于候选药物的后期细化。 |
| 基于量子化学的方法 ( 第一原理电子结构模拟 ) | 提供对分子相互作用、电子属性和反应机制的高度准确预测。 | 计算成本极高,无法随系统大小进行扩展,并且需要大量专业知识。 | 用于预测相互作用能、优化领先化合物以及了解原子层面的反应机制。 |
在实践中,研究人员通常使用分层策略,即廉价的高吞吐量预言机 (如快速计算屏幕) 过滤大量 AI 生成的分子。然后,使用更高准确性的预言机 (详细的模拟或实际实验) 评估最有前景的候选项。这可以将昂贵的实验室工作集中在最重要的 AI 建议上,从而节省时间和资源。
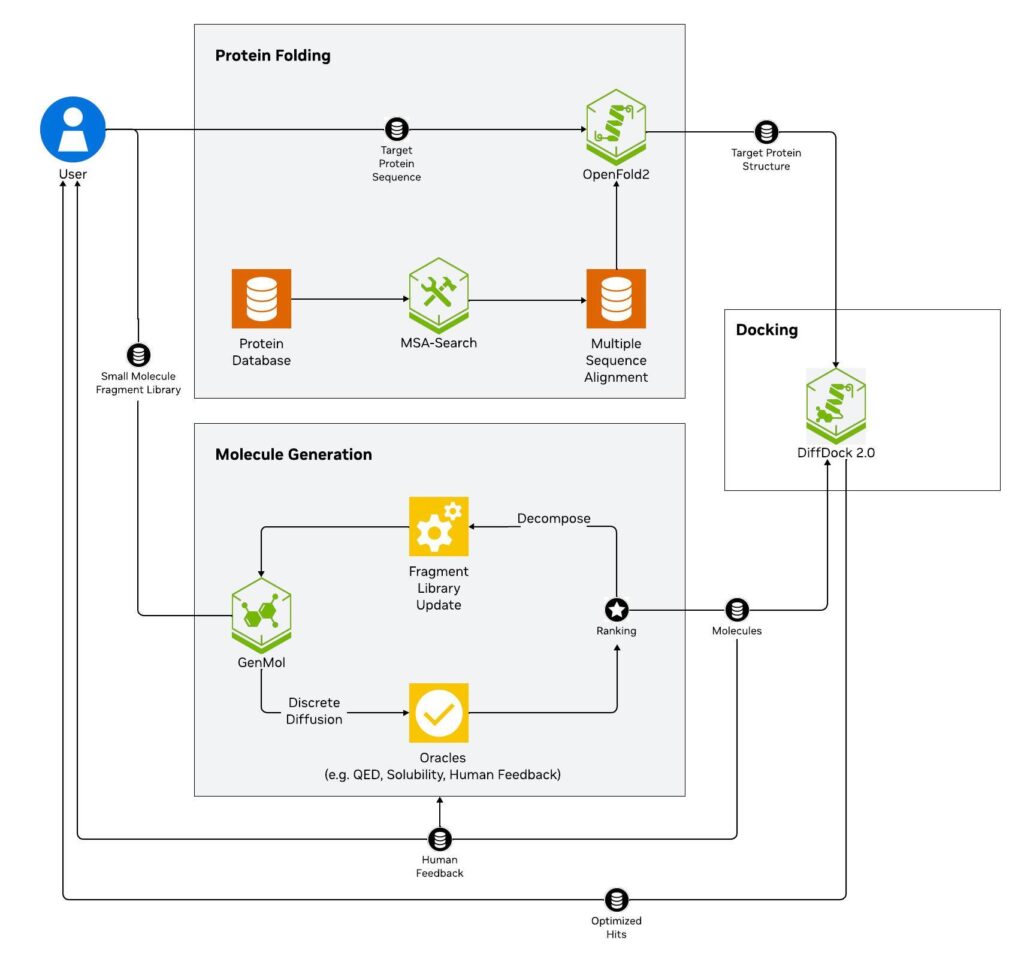
图 1。用于生成式虚拟筛选的 NVIDIA BioNeMo Blueprint 是用于 Oracle 引导分子设计和筛选的生成式 AI 示例
用于生成式虚拟筛选的 NVIDIA BioNeMo Blueprint (图 1) 就是一个示例:
-
目标蛋白质序列传递给 OpenFold2 NIM,后者使用 MSA-Search NIM 的多序列对齐准确确定蛋白质的 3D 结构。
-
将初始化化学库分解为碎片,并传递给 GenMol NIM,从而生成各种小分子。
-
对初始结构进行计算评分,并针对药物的多个特征 (例如预测溶解度和 Quantitative Estimate of Drug-likeness (QED)) 进行排名。
-
这些分数用于在迭代生成周期中对生成的分子进行排序和筛选,直到它们达到进一步测试所需的阈值。使用 DiffDock NIM 预测这些生成的小分子与目标蛋白的结合姿势。
-
最后,优化后的分子返回给用户进行合成和进一步的实验室验证。
按照以下伪代码,使用 NVIDIA GenMol NIM 实现迭代分子生成和优化流程。 此过程包括从片段库生成分子、使用 oracle 对其进行评估、选择 top 候选分子、将其分解为新的片段,以及重复该循环。
# Import necessary modules
from genmol import GenMolModel, SAFEConverter # Hypothetical GenMol API
from oracle import evaluate_molecule # Hypothetical oracle function
from rdkit import Chem
from rdkit.Chem import AllChem, BRICS
import random
# Define hyperparameters
NUM_ITERATIONS = 10 # Number of iterative cycles
NUM_GENERATED = 1000 # Number of molecules generated per iteration
TOP_K_SELECTION = 100 # Number of top-ranked molecules to retain
SCORE_CUTOFF = -0.8 # Example binding affinity cutoff for filtering
# Initialize GenMol model
genmol_model = GenMolModel()
# Load initial fragment library (list of SMILES strings)
with open('initial_fragments.smi', 'r') as file:
fragment_library = [line.strip() for line in file]
# Iterative molecule design loop
for iteration in range(NUM_ITERATIONS):
print(f"Iteration {iteration + 1} / {NUM_ITERATIONS}")
# Step 1: Generate molecules using GenMol
generated_molecules = []
for _ in range(NUM_GENERATED):
# Randomly select fragments to form a SAFE sequence
selected_fragments = random.sample(fragment_library, k=random.randint(2, 5))
safe_sequence = SAFEConverter.fragments_to_safe(selected_fragments)
# Generate a molecule from the SAFE sequence
generated_mol = genmol_model.generate_from_safe(safe_sequence)
generated_molecules.append(generated_mol)
# Step 2: Evaluate molecules using the oracle
scored_molecules = []
for mol in generated_molecules:
score = evaluate_molecule(mol) # Example: docking score, ML predicted affinity
scored_molecules.append((mol, score))
# Step 3: Rank and filter molecules based on oracle scores
scored_molecules.sort(key=lambda x: x[1], reverse=True) # Sort by score (higher is better)
top_molecules = [mol for mol, score in scored_molecules[:TOP_K_SELECTION] if score >= SCORE_CUTOFF]
print(f"Selected {len(top_molecules)} high-scoring molecules for next round.")
# Step 4: Decompose top molecules into new fragment library
new_fragment_library = set()
for mol in top_molecules:
# Decompose molecule into BRICS fragments
fragments = BRICS.BRICSDecompose(mol)
new_fragment_library.update(fragments)
# Step 5: Update fragment library for next iteration
fragment_library = list(new_fragment_library)
print("Iterative molecule design process complete.")
按照以下伪代码,使用 MolMIM NIM 实现由 Oracle 驱动的迭代分子生成过程。此方法包括生成分子、使用 Oracle 评估分子、选择出色的候选分子,以及根据 Oracle 反馈完善生成过程 。
# Import necessary modules
from molmim import MolMIMModel, OracleEvaluator # Hypothetical MolMIM and Oracle API
import random
# Define hyperparameters
NUM_ITERATIONS = 10 # Number of iterative cycles
NUM_GENERATED = 1000 # Number of molecules generated per iteration
TOP_K_SELECTION = 100 # Number of top-ranked molecules to retain
SCORE_CUTOFF = 0.8 # Example oracle score cutoff for filtering
# Initialize MolMIM model and Oracle evaluator
molmim_model = MolMIMModel()
oracle_evaluator = OracleEvaluator()
# Iterative molecular design loop
for iteration in range(NUM_ITERATIONS):
print(f"Iteration {iteration + 1} / {NUM_ITERATIONS}")
# Step 1: Generate molecules using MolMIM
generated_molecules = molmim_model.generate_molecules(num_samples=NUM_GENERATED)
# Step 2: Evaluate molecules using the oracle
scored_molecules = []
for mol in generated_molecules:
score = oracle_evaluator.evaluate(mol) # Returns a score between 0 and 1
scored_molecules.append((mol, score))
# Step 3: Rank and filter molecules based on oracle scores
scored_molecules.sort(key=lambda x: x[1], reverse=True) # Sort by score (higher is better)
top_molecules = [mol for mol, score in scored_molecules[:TOP_K_SELECTION] if score >= SCORE_CUTOFF]
print(f"Selected {len(top_molecules)} high-scoring molecules for next round.")
# Step 4: Update MolMIM model with top molecules
molmim_model.update_model(top_molecules)
print("Iterative molecular design process complete.")
将 Oracle(基于实验和计算的反馈机制)集成到 AI 驱动的分子设计中,从根本上改变了药物设计。通过在生成模型和现实世界的验证之间建立连续循环,研究人员可以超越理论分子生成,转而使用实用、可合成且具有功能性的候选药物。
这种实验室在环方法能够:
-
使用 GenMol NIM 和 MolMIM NIM 等 AI 模型加快迭代周期,根据实验或高精度计算反馈生成和完善分子。
-
高效的资源分配,在这种情况下,计算预言机可以快速筛选数千种分子,然后再将成本高昂的实验室实验重点放在具有广阔前景的候选分子上。
-
通过将真实的实验结果整合到 AI 模型中,帮助他们更好地预测药物相似性,从而提高准确性和泛化程度。
随着 AI 模型和 Oracle 系统变得更加先进,我们即将进入 AI 和实验科学共同发展的时代,推动药物设计取得突破性进展。通过集成高质量的 Oracle,虚拟分子设计与现实世界成功之间的差距将继续缩小,为精准医疗等领域带来新的可能性。





相关贴子
-
 技术分享
技术分享选 CPU 看核心数量还是时钟速度?
2024.10.11 27分钟阅读 -
 技术分享
技术分享【速看】TensorFlow 2.17 发行说明
2024.11.01 20分钟阅读 -
 技术分享
技术分享Ollama 与 vLLM 深度对比:大模型部署方案如何选型
2026.05.18 70分钟阅读